Atròfia muscular espinal en nens
L'atròfia muscular espinal és una patologia greu que sovint fa accions senzilles, com ara caminar, estar asseguda i inaccessible per a un nen. El nadó pot estar privat de fins i tot una oportunitat tan natural com la respiració independent. És molt difícil fer previsions per a aquesta patologia, Al cap ia la fi, no existeix cap tractament ni profilaxi especial, però tot depèn de la forma de la malaltia i dels factors, quina medicina no pot trobar una explicació.
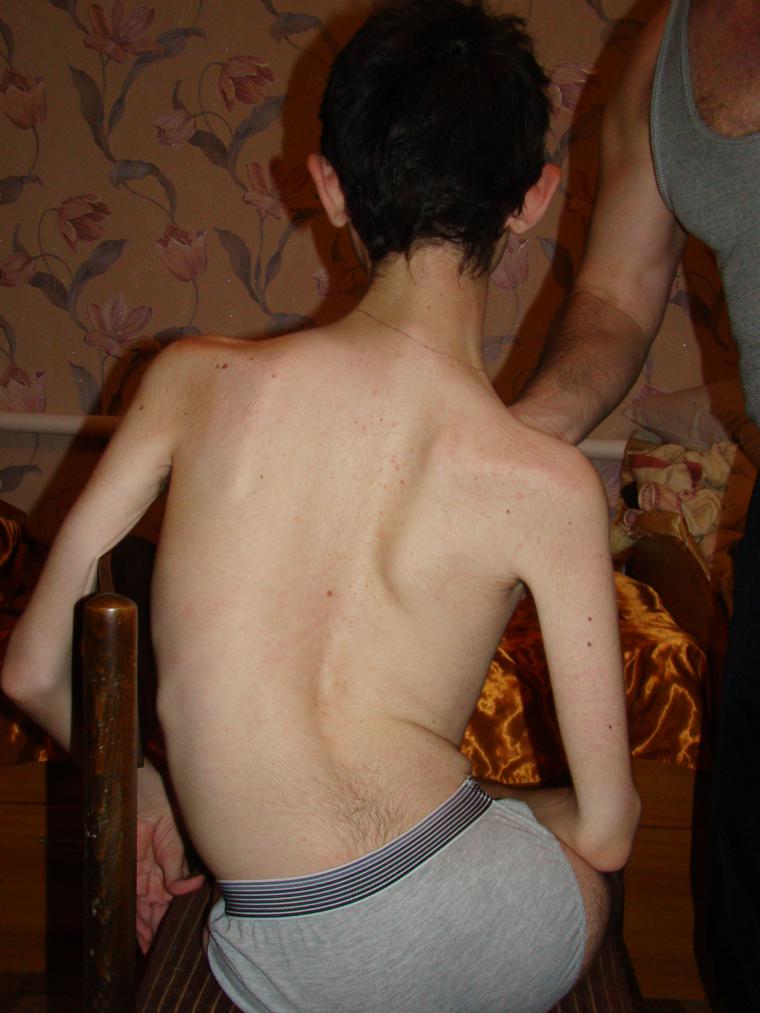
Què és això?
Parlant d’atròfia muscular espinal, implica no una sola malaltia específica, sinó tot un grup de malalties sota l’abreviatura general CMA. Tots ells són hereditaris i estan associats amb la degeneració de les cèl·lules nervioses de la medul·la espinal, que són responsables de les funcions motores.
Entre els patologies genètiques en nens Les atròfies musculars espinals ocupen llocs principals en termes de freqüència de propagació. I aproximadament un de cada sis mil nens neixen amb un diagnòstic tan terrible. En el 50% dels casos, els nens no viuen fins als dos anys i moren. La resta de la vida és una discapacitat.

El problema, segons els genetistes, és molt més ampli del que sembla de les estadístiques donades.
La malaltia es desenvolupa a causa de la mutació de certs gens i un d’ells és SMN1, que es considera el principal "culpable" de la patologia, en una forma modificada de present recessiu a cada cinquanta habitant del planeta. Això vol dir que els pares sans, que ni tan sols s'adonen que són portadors recessius del gen mutat, poden tenir un bebè amb atròfia muscular espinal.
El grup de malalties es va descriure per primera vegada al segle XIX per Guido Verding, el nom del qual es va nomenar més tard una de les varietats infantils d'AGR.

Classificació
La forma més comuna de SMA en nens és proximal. Està representat per diversos tipus de malalties, que no es manifesten immediatament després del naixement del nen.
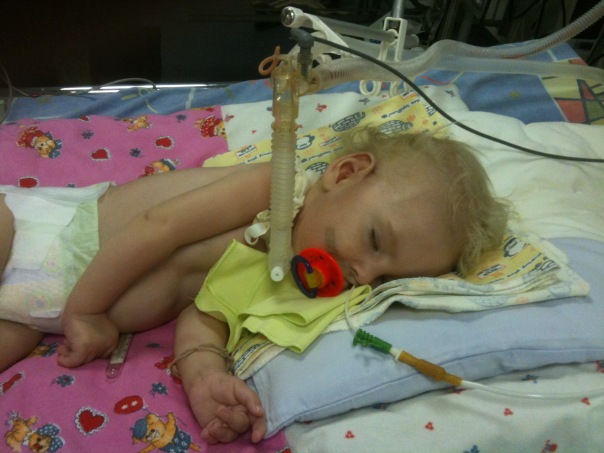
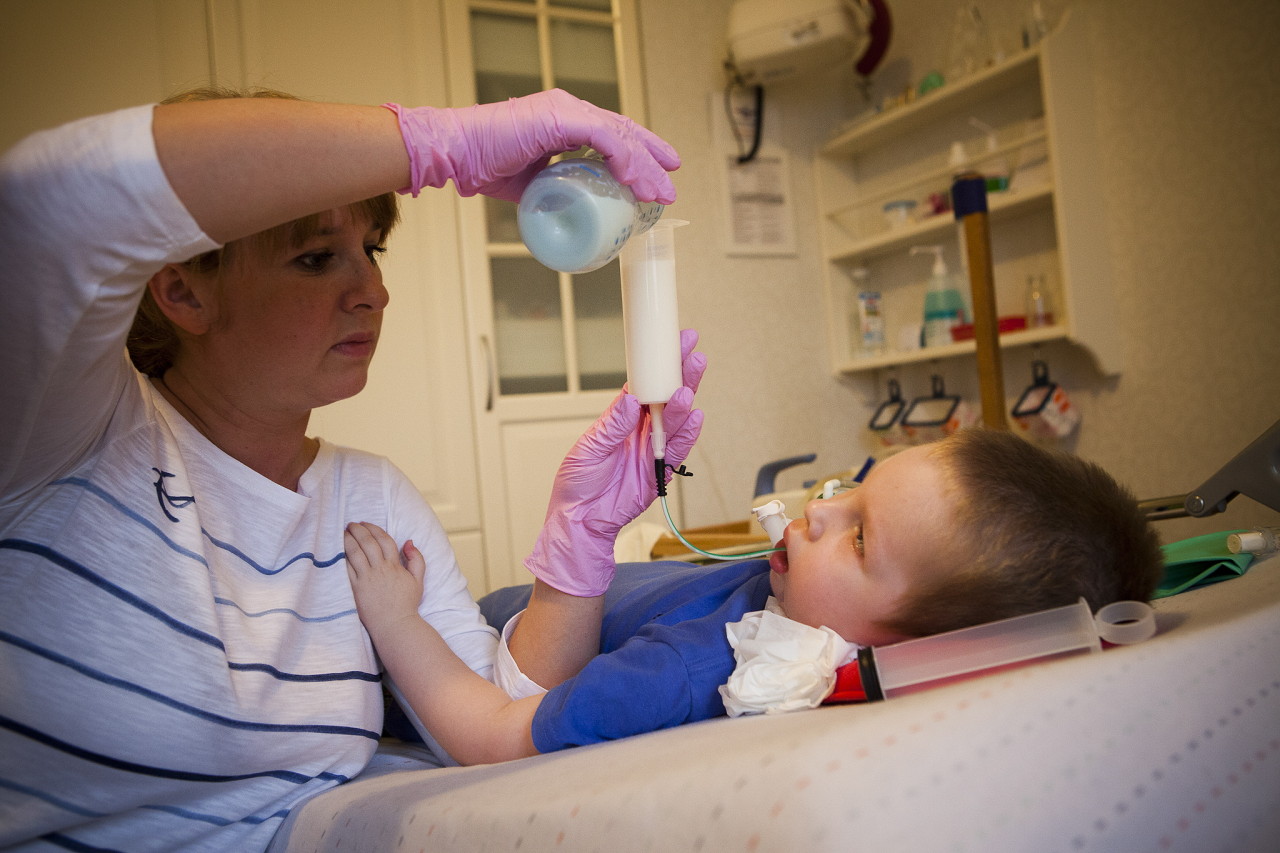
- Malaltia de Verding-Hoffman - MCA de tipus 1, malalties infantils severesque es manifesta en els primers sis mesos de la vida del nen. Les previsions amb ella són les més desfavorables, la majoria dels pacients moren. Un nen amb SMA de tipus 1 no pot suportar-se ni seure ni desplaçar-se de manera independent. Molts nounats tenen problemes de reflexió per succió i per empassar. Sovint no hi ha possibilitat de respiració espontània o de respiració difícil.
- Atrophy Dubovitsa - CMA type 2, infants tardans. Normalment es produeix entre els sis mesos i un any i mig més tard. El nen no pot caminar, posar-se de peu, però és capaç de seure, el menjar no és pertorbat, fa front a la tasca de deglutir, xuclar. El temps de vida del bebè depèn de l'estat dels músculs respiratoris.
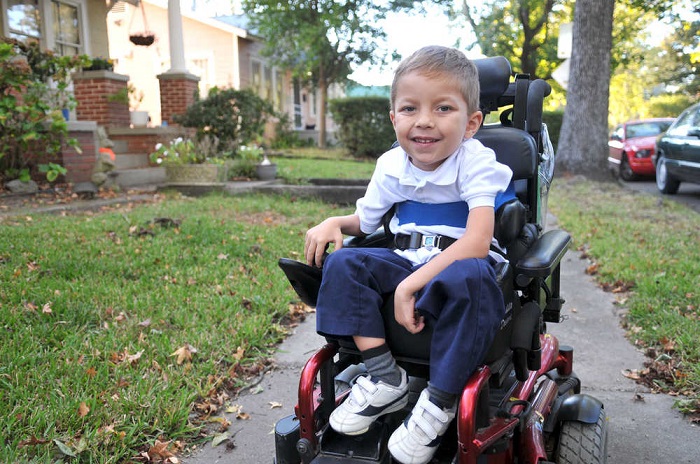
- Atròfia dels tipus Kugelberg-Welander - CMA 3, infantil. Normalment es troba en l'edat d'un any i mig, normalment en dos anys. Forma més favorable del pronòstic. Els pacients petits poden estar de peu, seure, moure's, però experimenten una gran debilitat i, per tant, en la majoria dels casos necessiten una cadira de rodes, sense la qual cosa la seva activitat vital normal és difícil.
- Atròfia de Kennedy - CMA tipus 4, bulbospinalnaya. Normalment es considera una forma adulta, però ocasionalment es detecta en nens després de 15 anys. La influència de la vida poques vegades es veu afectada, el debilitament dels músculs es produeix lentament, gradualment, una persona que portava una vida normal i es considerava sana, esdevindria discapacitada i perd la capacitat de moure's de manera independent.


L'atròfia de primera mà més o menys coneguda de Duchenne i l'atròfia de Vulpian són "adults" SMA, el primer sol detectar-se després de 18 anys i el segon després de 20 anys.
Als nens, no només es registren formes aïllades de SMA, quan res no es molesta excepte la distròfia muscular, però també forma combinada, quan l'atròfia vertebral no és l'únic diagnòstic i el nen té altres problemes genètics o congènits, com ara defectes cardíacs i vasculars, oligofrenia.
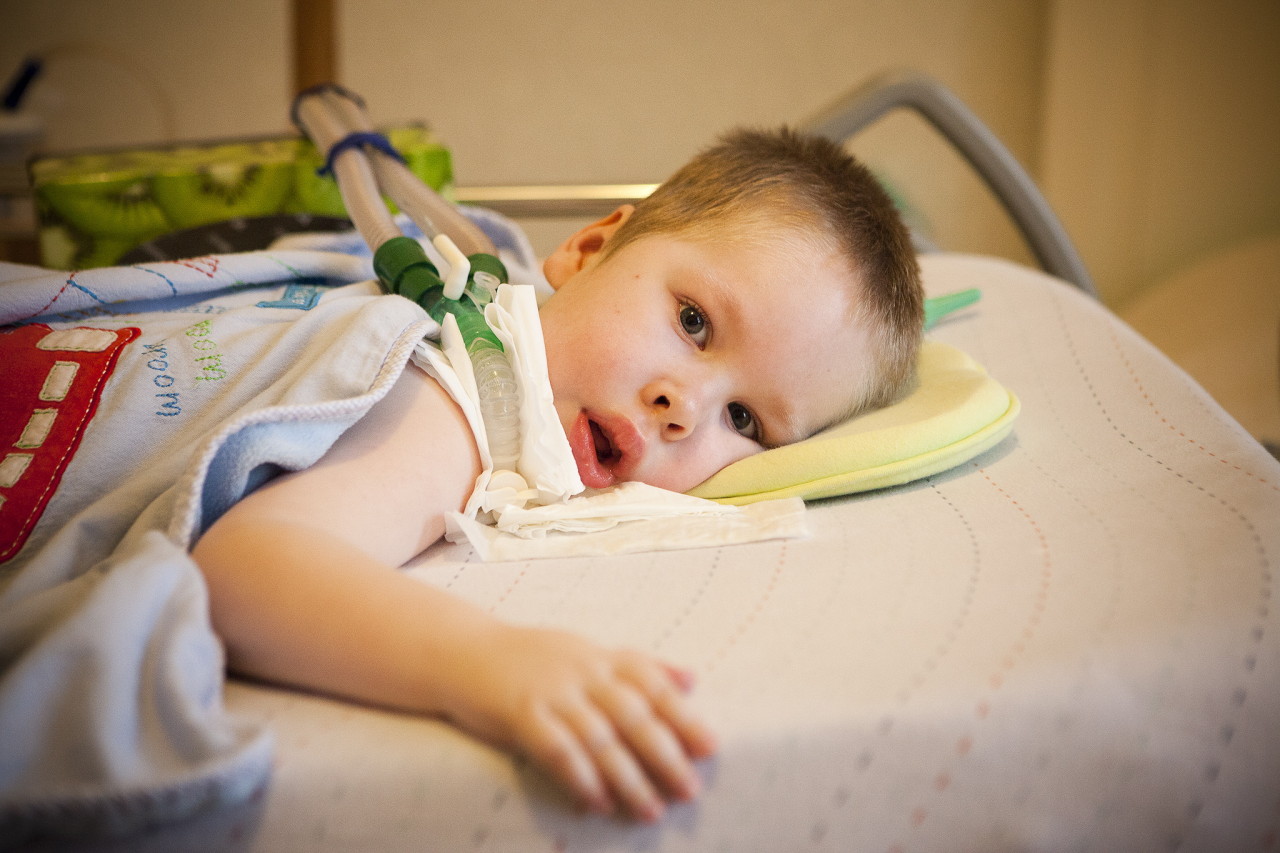
Raons
Com ja s'ha dit, estem parlant d'una malaltia genètica i, per tant, els motius de la seva aparició són el camp de la recerca de genètics. El nen hereta un dels gens recessius del cinquè cromosoma (aquests poden ser els gens SMN, NAIP, H4F5, BTF2p44).
La probabilitat de transmetre aquest gen a una descendència d’un portador és elevada: un 25%. Si tant la mare com el pare són portadors ocults del gen mutat, llavors la probabilitat d’AGR en un nen és del 50%. El gen anormal afectat impedeix la producció normal de proteïnes SMN i les cèl·lules nervioses responsables de les funcions motores dels músculs de la medul·la espinal comencen a morir gradualment. El procés de la seva mort continua fins i tot després que neixi el bebè.
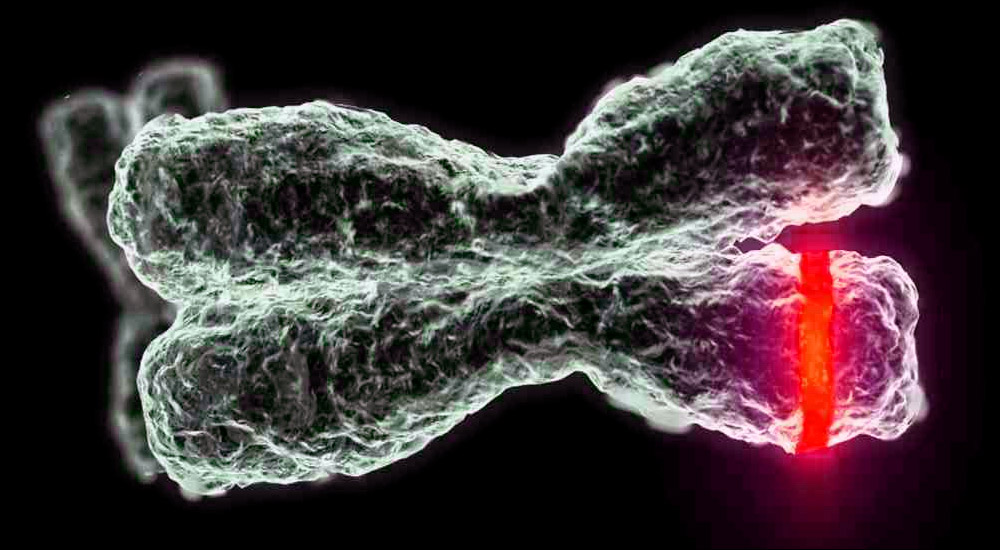

Manifestacions
Els símptomes depenen del tipus de malaltia. Com que considerem només quatre tipus de nens, cal assenyalar que la debilitat muscular i l'atròfia muscular són característiques de tots. En cas contrari, cada tipus té el seu propi quadre clínic i característiques distintives.
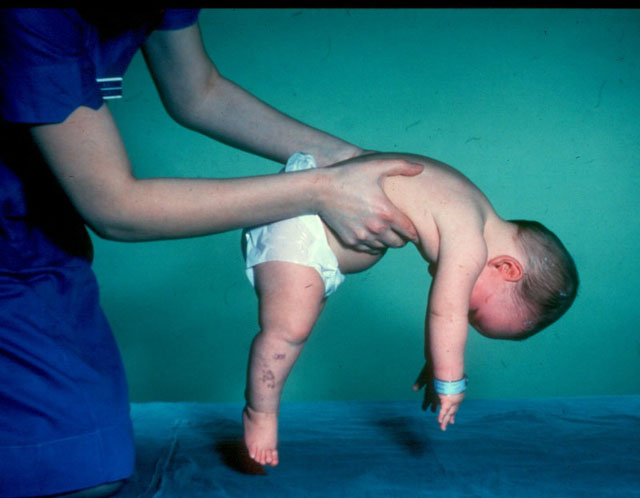
- CMA tipus 1 (atrofia Verding-Hoffman) disponible per a la seva detecció fins i tot durant l’embaràs. El metge pot sospitar que la malaltia del fetus es remou molt lentament. Però per confirmar el diagnòstic en l'etapa de portar un nen és difícil, normalment es produeix després del part. Un noi amb tanta atròfia no pot aguantar el cap, llançar-se de costat a costat, no seure. Gairebé sempre es troba a l'esquena, la seva postura és relaxada, no aixeca les cames, no les uneix, no posa els palmells junts. En una etapa molt primerenca, pot haver-hi problemes enormes per alimentar el nen, ja que s'empassa molt malament o no funciona. La majoria dels nens moren abans dels dos anys. Alguns aconsegueixen viure fins a set o vuit anys, però l'atrofia només empitjora. Normalment la mort es produeix a causa de la insuficiència del cor, dels pulmons i dels òrgans digestius.
- Tipus CMA 2 (atrofia Dubovitsa) en néixer, normalment no es detecta, ja que el nen és capaç de respirar, d'empassar menjar, i només després de sis mesos es fa evident el progrés de l'atròfia muscular. Si els primers símptomes es produeixen a l'edat en què el nen ja ha après a situar-se al bressol, després un signe de tall de les cames, una gota irracional de les molles pot ser un signe brillant. A poc a poc, es fa difícil d'empassar. Amb el temps, el nen comença a necessitar una cadira de rodes.
- CMA tipus 3 (amyotrofia de Kügelberg-Welander) pot aparèixer a qualsevol edat després dels 2 anys d’edat. Un nen que normalment creix i es desenvolupa gradualment comença a queixar-se de debilitat, generalment a les espatlles, avantbraços. A mesura que avança, li fa difícil córrer, caminar per les escales, posar-se a la gatzoneta. Tot depèn de la cura: alguns conserven la capacitat de moure's de manera independent durant molts anys.
- CMA tipus 4 (atrofia Kennedy) es produeix només en pacients homes, ja que es considera vinculada al cromosoma sexual X. Els primers signes són debilitat a la regió dels músculs de la cuixa i els nervis cranials es veuen afectats gradualment. La malaltia progressa lentament.

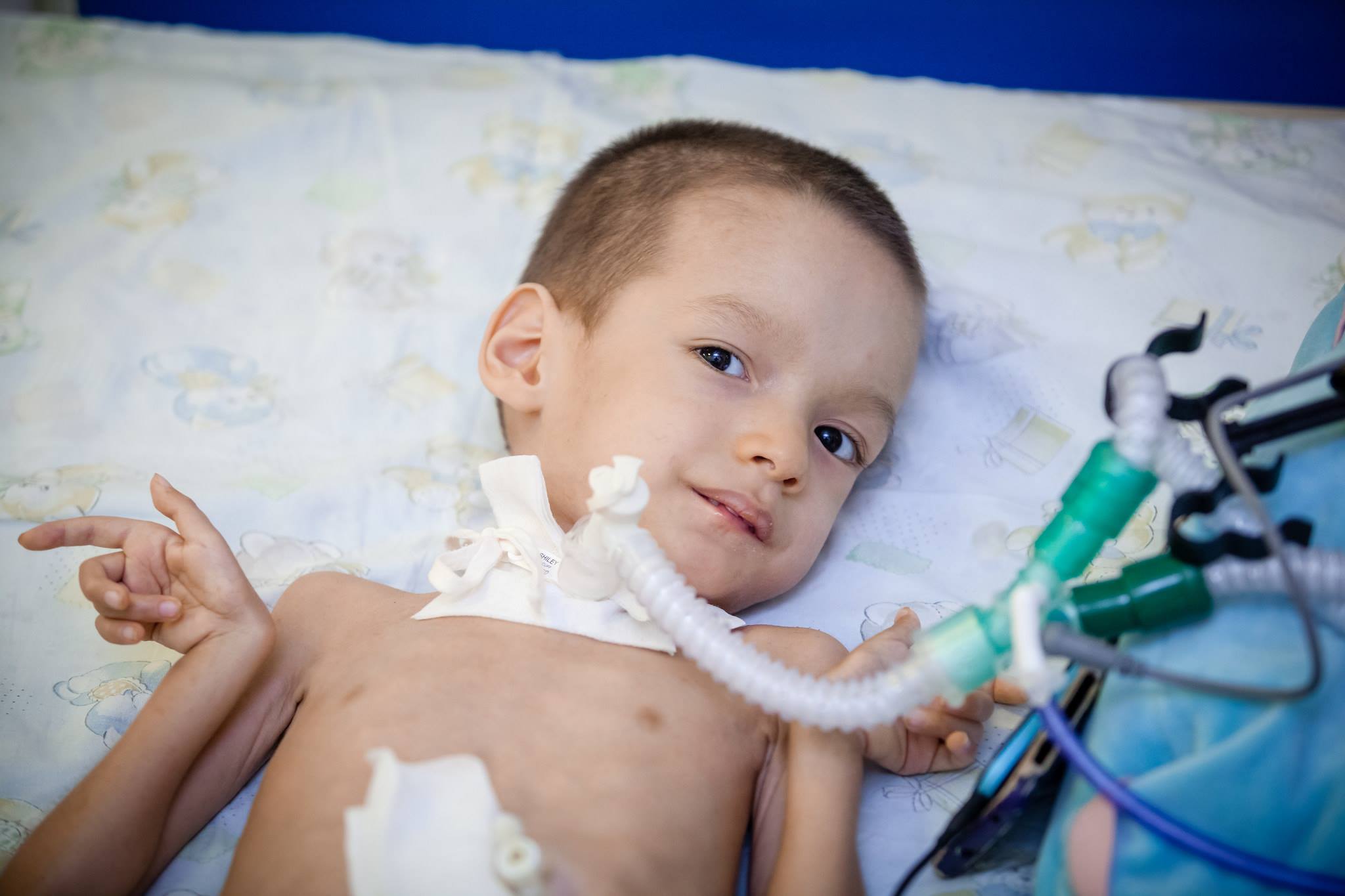
Tractament
Malauradament, avui dia la ciència mèdica no pot oferir mètodes i mitjans per al tractament de l’ASM. No hi ha aquests mètodes. Mantenir les funcions del cos i maximitzar el període fins que el nen es pugui moure, medicaments com Prozerin, Oksazil. Redueixen l’activitat d’un enzim capaç d’escindir acetilcolina, que transmet un pols d’excitació a través de les fibres del sistema nerviós.
També es recomana ús sistemàtic de fàrmacs que milloren el metabolisme energètic a nivell cel·lular, vitamines del grup B, medicaments nootròpics, així com preparats de potassi i àcid nicotínic.
Un nen amb SMA mostra una dieta alta en proteïnesPerò estudis recents han demostrat que el paper de la dieta és una mica exagerat: no hi ha proves que un alt contingut de proteïnes en els aliments afecti almenys d'alguna manera la taxa de progressió de la malaltia.
Però amb les calories s’ha de tenir més cura: a causa de la reducció de l’activitat muscular, el nen pot guanyar lliures extra.
Ajudar a allargar el període d’una vida més o menys satisfactòria ajudarà massatge terapèutic, UHF, electroforesi, exercicis de respiració per al manteniment dels músculs respiratoris, natació. Es recomana utilitzar aparells ortopèdics espinals o toràcics de suport.
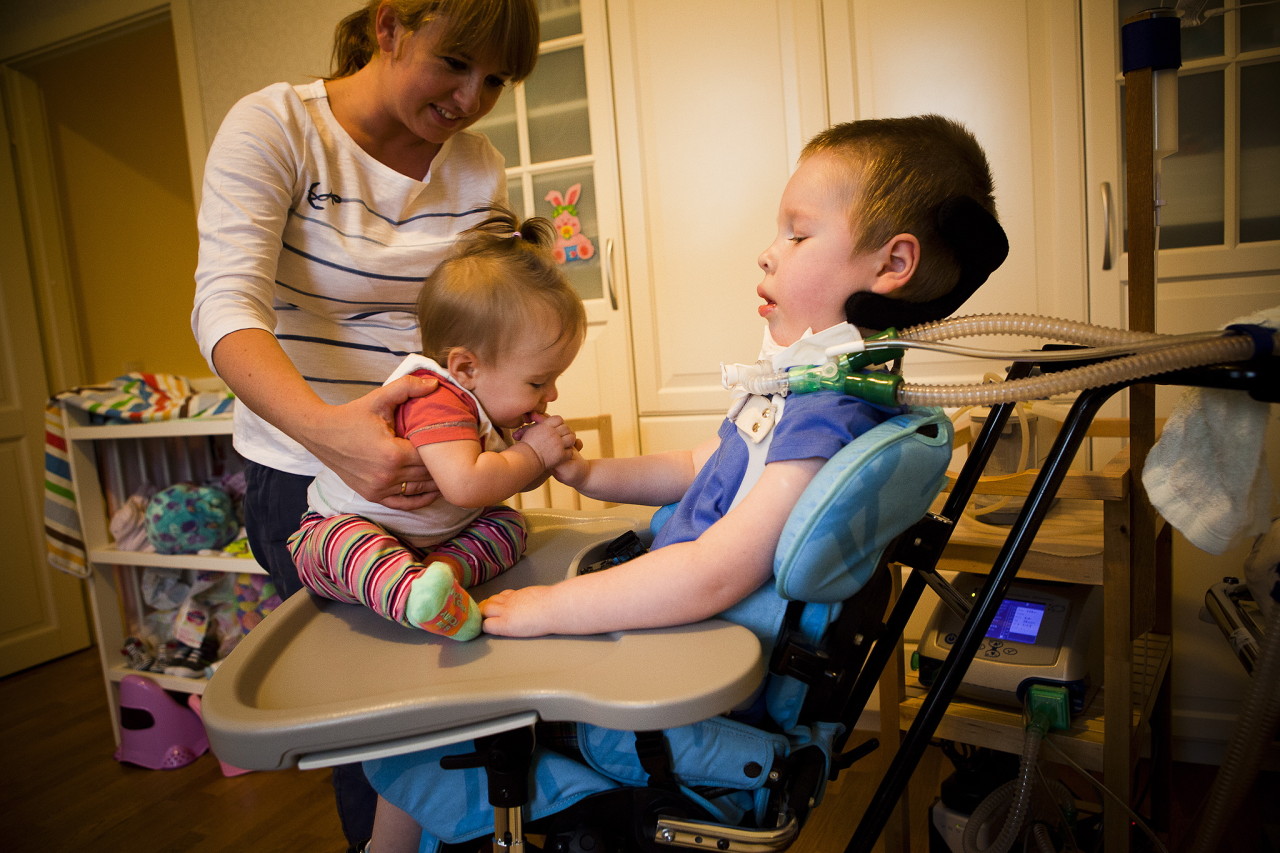

Més informació sobre la malaltia li diu a un especialista el vídeo que apareix a continuació.