Spinal muskelatrofi hos barn
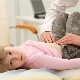
Spinal muskulær atrofi er en alvorlig patologi som ofte gjør enkle handlinger, for eksempel å gå, sitte, utilgjengelig for et barn. Barnet kan bli fratatt selv en så naturlig mulighet som uavhengig pust. Det er veldig vanskelig å lage spådommer for denne patologien, Tross alt eksisterer det ingen spesiell behandling eller profylakse, men alt avhenger av formen og lidelsen, hvilken medisin kan ikke finne en forklaring.
Hva er dette?
Når det gjelder spinal muskulær atrofi, betyr ikke en bestemt sykdom, men en hel gruppe sykdommer under den generelle forkortelsen CMA. Alle er arvelige og er forbundet med degenerasjon av nervecellene i ryggmargen, som er ansvarlige for motorfunksjonene.
Blant genetiske patologier hos barns spinal muskulære atrofier opptar ledende steder når det gjelder frekvens av spredning. Og om en av seks tusen barn er født med en så forferdelig diagnose. I 50% av tilfellene lever barn ikke i en alder av to og dør. Resten av livet er et funksjonshemning.

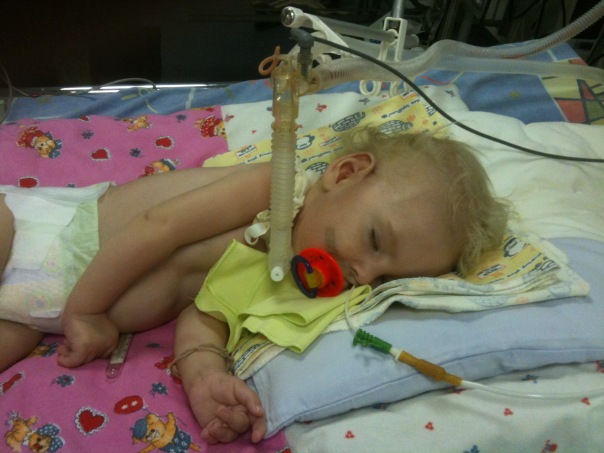
Problemet, ifølge genetikere, er mye bredere enn det kan virke ut fra statistikken gitt.
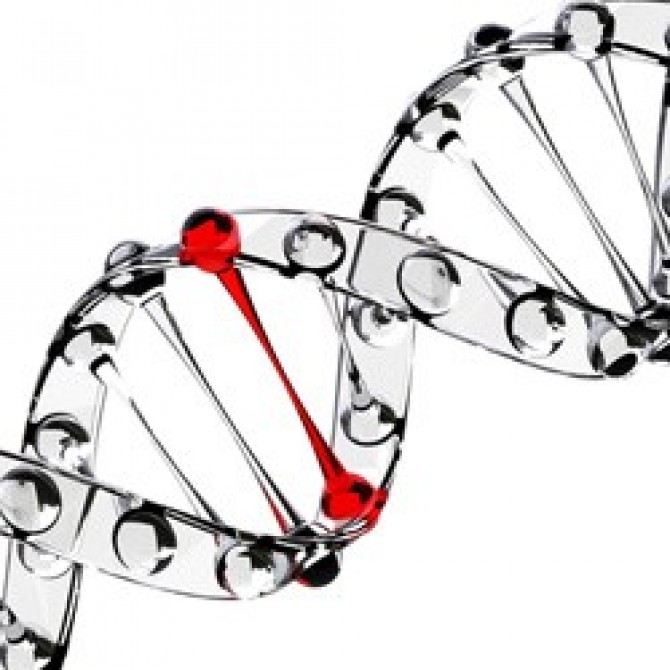
Sykdommen utvikler seg på grunn av mutasjonen av visse gener, og en av dem er SMN1, som betraktes som den viktigste "skyldige" av patologi, i en modifisert form av resessivt tilstede i hver femtiende innbygger på planeten. Dette betyr at friske foreldre, som ikke engang innser at de er recessive bærere av det muterte genet, kan ha en baby med spinal muskulær atrofi.
Gruppen av sykdommer ble først beskrevet i det 19. århundre av Guido Verding, der navnet senere ble kåret til en av barnas varianter av AGR.

klassifisering
Den vanligste formen for SMA hos barn er proksimal. Det er representert av flere typer sykdommer, og ikke alle blir tydelige umiddelbart etter barnets fødsel.
- Verding-Hoffman sykdom - Type 1 MCA, alvorlig spedbarnssykdomsom manifesterer seg i de første seks månedene av barnets liv. Prognoser med hennes mest ugunstige, de fleste pasienter dør. Et barn med type 1 SMA kan hverken stå eller sitte, eller rulle over uavhengig. Mange nyfødte har nedsatt sug og svelger reflekser. Ofte er det ingen mulighet for spontan pust eller å puste er vanskelig.
- Atrofi Dubovitsa - CMA type 2, sent spedbarn. Det oppstår vanligvis mellom seks måneder og et og et halvt år senere. Barnet kan ikke gå, stå, men er i stand til å sitte, maten er ikke forstyrret, han klarer å svelge, suger. Hvor lenge babyen skal leve, avhenger av tilstanden til luftveiene.
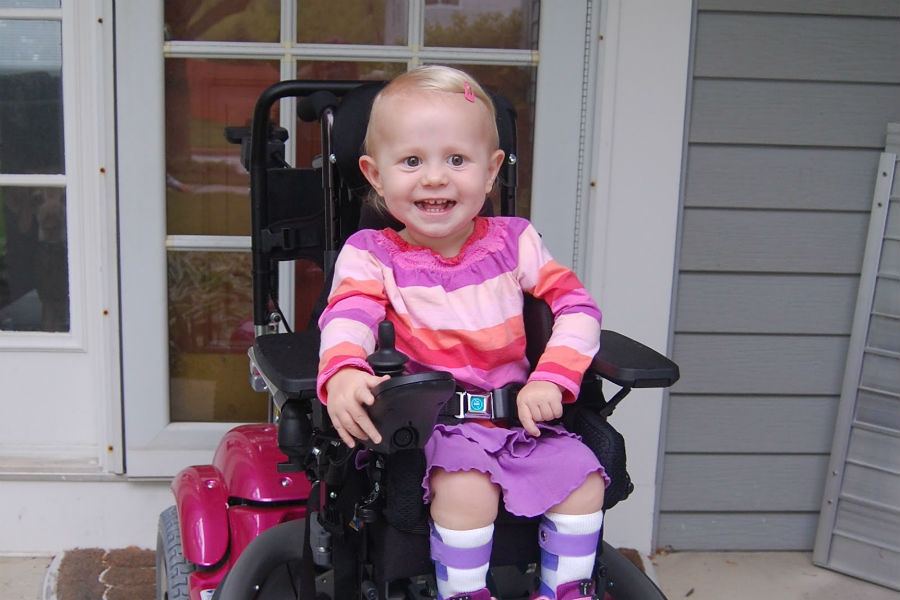
- Atrofi av Kugelberg-Welander - CMA 3 typer, infantile. Vanligvis funnet i en og ett halvt år, vanligvis om to år. Prognostisk mer gunstig form. Små pasienter kan stå, sitte, flytte, men oppleve stor svakhet, og de trenger derfor i de fleste tilfeller en rullestol, uten hvilken deres normale livsviktige aktivitet er vanskelig for dem.
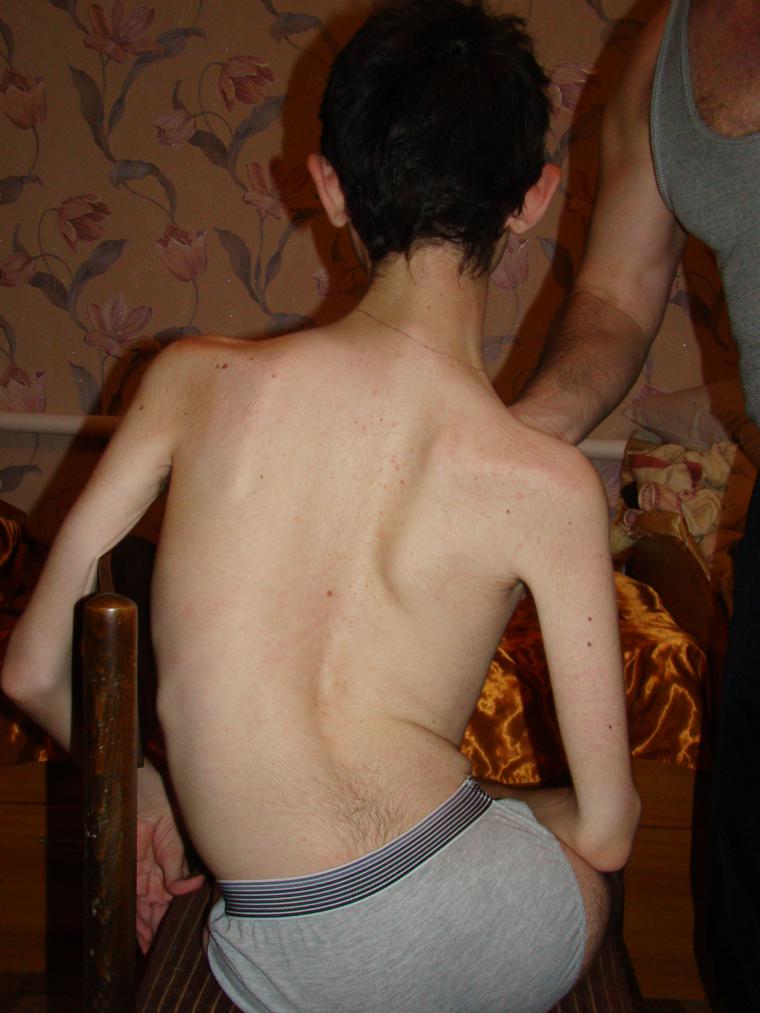
- Kennedy atrofi - CMA type 4, bulbospinalnaya. Vanligvis betraktes som en voksenform, men av og til oppdaget hos barn etter 15 år. Innflytelse av liv er sjelden påvirket, svekkelse av muskler skjer sakte, gradvis, en person som ledet et normalt liv og ansett seg frisk, blir til slutt deaktivert og mister evnen til å bevege seg selvstendig.


Mer eller mindre kjent førstegangsatrofi av Duchenne og atrofi av Vulpian er SMA "voksne", den første er vanligvis oppdaget etter 18 år og den andre etter 20 år.
Hos barn registreres ikke bare isolerte former for SMA, når det ikke er noe annet enn muskeldystrofi, men også kombinerte former når spinalatrofi ikke er den eneste diagnosen, og barnet har andre genetiske eller medfødte problemer, som for eksempel hjerte og vaskulære defekter, oligofreni.
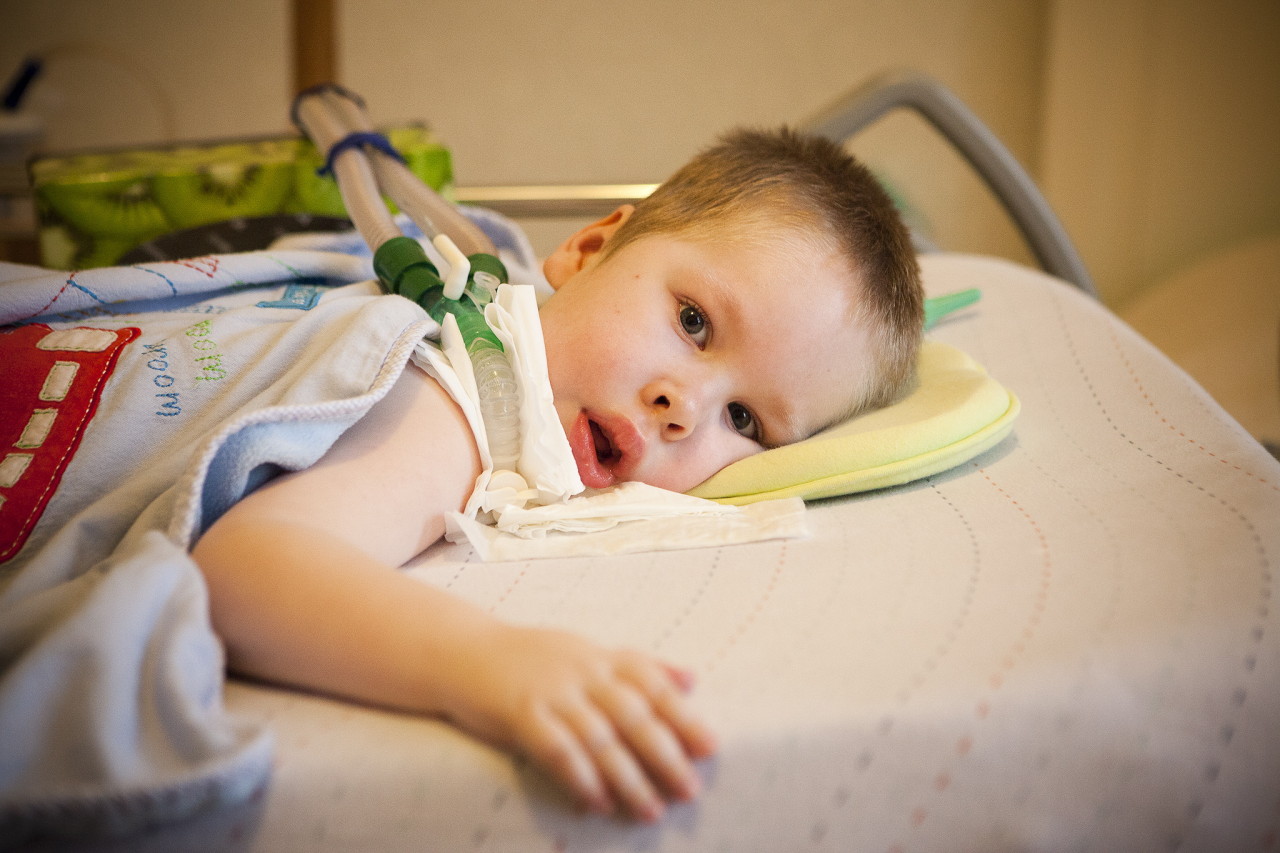
årsaker
Som allerede nevnt, snakker vi om en genetisk sykdom, og derfor er årsakene til forekomsten hennes å søke etter genetikere. Barnet arver et av de recessive gener på det femte kromosomet (disse kan være SMN, NAIP, H4F5, BTF2p44-gener).
Sannsynligheten for å overføre et slikt gen til et avkom fra en bærer er høy - 25%. Hvis både mor og pappa er skjulte bærere av det muterte genet, er sannsynligheten for AGR hos et barn 50%. Det berørte unormale genet forhindrer normal SMN proteinproduksjon og nervecellene som er ansvarlige for motorfunksjonene i muskler i ryggmargen begynner å dø gradvis. Prosessen med deres død fortsetter selv etter at barnet er født.
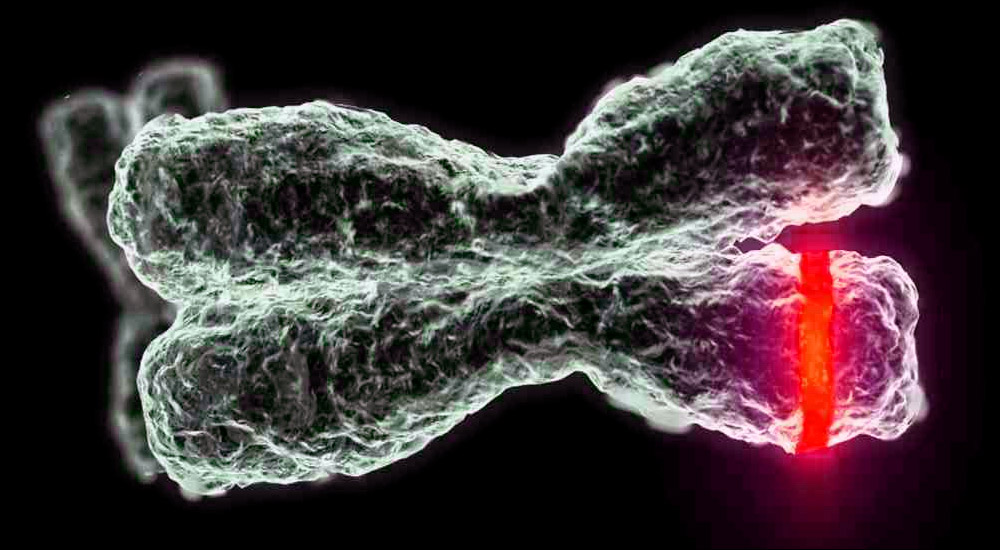

manifestasjoner
Symptomer avhenger av sykdommens type. Siden vi bare ser på fire typer barn, bør det bemerkes at muskelsvakhet og muskelatrofi er karakteristisk for alle. Ellers har hver type sitt eget kliniske bilde og særegne egenskaper.
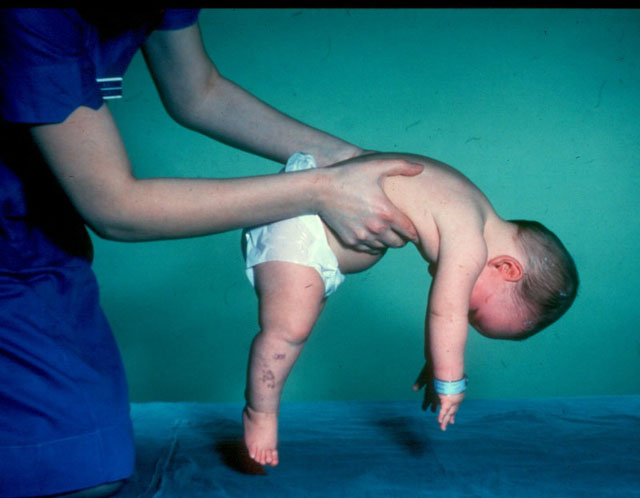
- CMA type 1 (Verding-Hoffman atrofi) tilgjengelig for deteksjon selv under graviditet. Legen kan mistenke sykdommen i fosteret med svært treg omrøring. Men for å bekrefte diagnosen på scenen for å bære et barn er vanskelig, skjer det vanligvis etter fødselen. Et barn med slik atrofi kan ikke holde hodet selv, kaste fra side til side, han setter seg ikke ned. Han ligger nesten alltid på ryggen, hans stilling er avslappet, han løfter ikke bena, bringer dem ikke sammen, legger ikke håndflatene sammen. På et veldig tidlig stadium kan det være store problemer for å mate barnet, fordi han svelger det viser seg veldig dårlig eller ikke fungerer. De fleste barn dør før to år. Noen klarer å leve i syv eller åtte år, men atrofi blir bare verre. Vanligvis kommer døden på grunn av mangel på hjerte, lunger, fordøyelseskanaler.
- CMA 2 type (atrofi Dubovitsa) ved fødselen blir det vanligvis ikke oppdaget, fordi barnet er i stand til å puste, svelge mat, og først etter seks måneder blir fremdriften av muskelatrofi tydelig. Hvis de første symptomene oppstår i en alder når barnet allerede har lært å stå i krybben, så et skjæremerke på beina, kan en urimelig dråpe av krummer være et lystegn. Gradvis blir det vanskelig å svelge. Over tid begynner barnet å trenge en rullestol.
- CMA type 3 (amyotrofi av Kügelberg-Welander) kan dukke opp i alle aldre etter 2 år. Et barn som normalt vokste og utviklet seg, begynner å klage av svakhet, vanligvis i skuldrene, underarmene. Når det skrider frem, blir det vanskelig for ham å løpe, gå trapper, knep. Alt avhenger av omsorg - noen beholder evnen til å bevege seg selvstendig i mange år.
- CMA type 4 (Kennedy atrofi) forekommer bare hos mannlige pasienter, siden det anses å være knyttet til sexkromosomet X. De første tegnene er svakhet i lårmuskelområdet, og kraniale nerver blir gradvis påvirket. Sykdommen utvikler seg sakte.

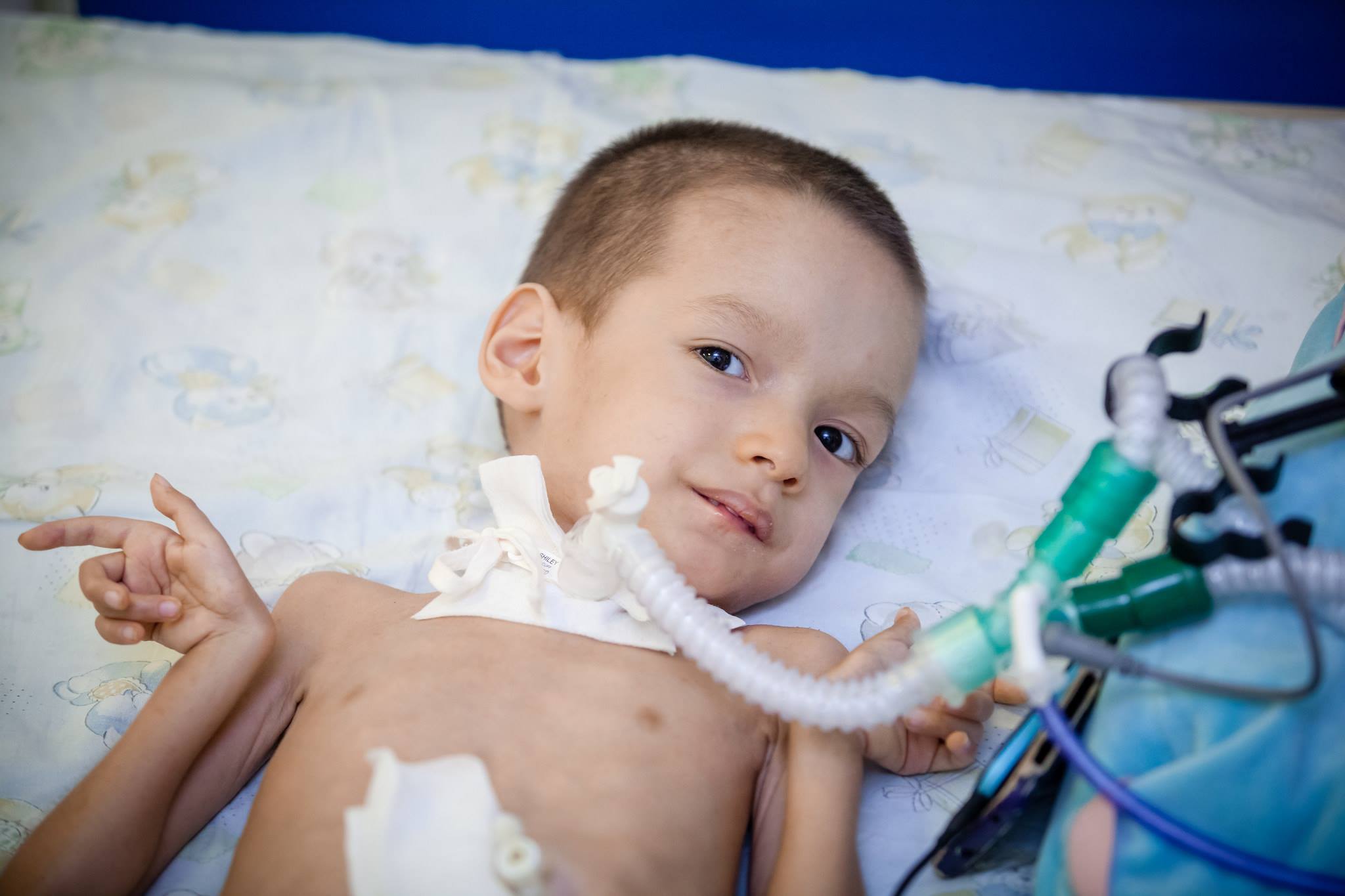
behandling
Dessverre, i dag kan medisinsk vitenskap ikke tilby metoder og midler til behandling av SMA. Det finnes ingen slike metoder. For å opprettholde kroppens funksjoner og maksimere perioden til barnet kan bevege seg, medisiner som Prozerin, Oksazil. De reduserer aktiviteten til et enzym som er i stand til å spalte acetylkolin, som overfører en eksitasjonspuls gjennom fibrene i nervesystemet.
Anbefales også systematisk bruk av narkotika som forbedrer energiomsetningen på mobilnivå, gruppe B-vitaminer, nootropiske stoffer, samt kalium- og nikotinsyrepreparater.
Et barn med SMA høyt protein diett vistMen nyere studier har vist at diettens rolle er litt overdrevet - det er ingen bevis for at høyt innhold av protein i maten i det minste på en eller annen måte påvirker sykdomsprogresjonen.
Men med kaloriene bør man være mer forsiktig - på grunn av redusert muskelaktivitet, kan barnet raskt få ekstra pounds.
Å hjelpe til med å forlenge perioden med et mer eller mindre tilfredsstillende liv, vil hjelpe terapeutisk massasje, UHF, elektroforese, pusteøvelser for vedlikehold av luftveiene, svømming. Det anbefales å bære støttende spinal- eller thoracale ortopediske apparater.
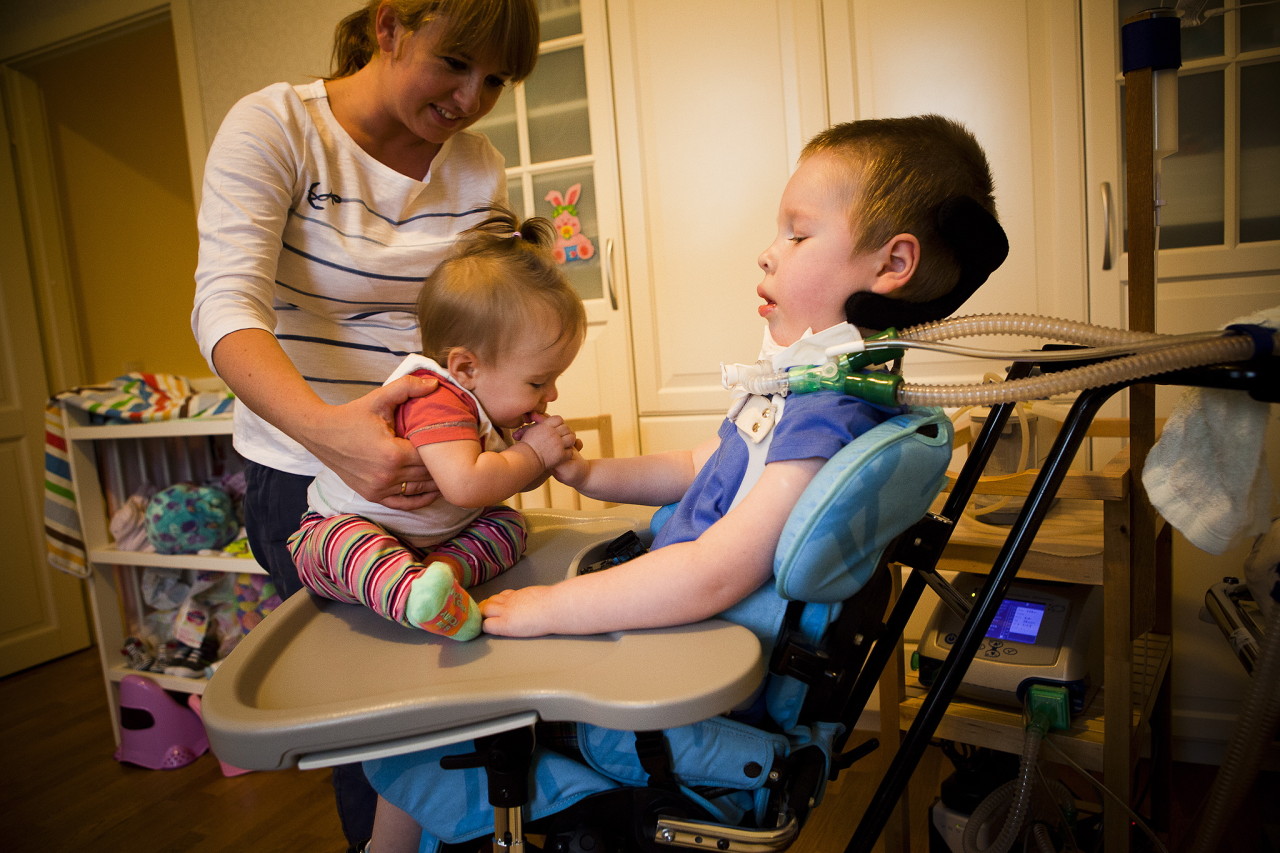
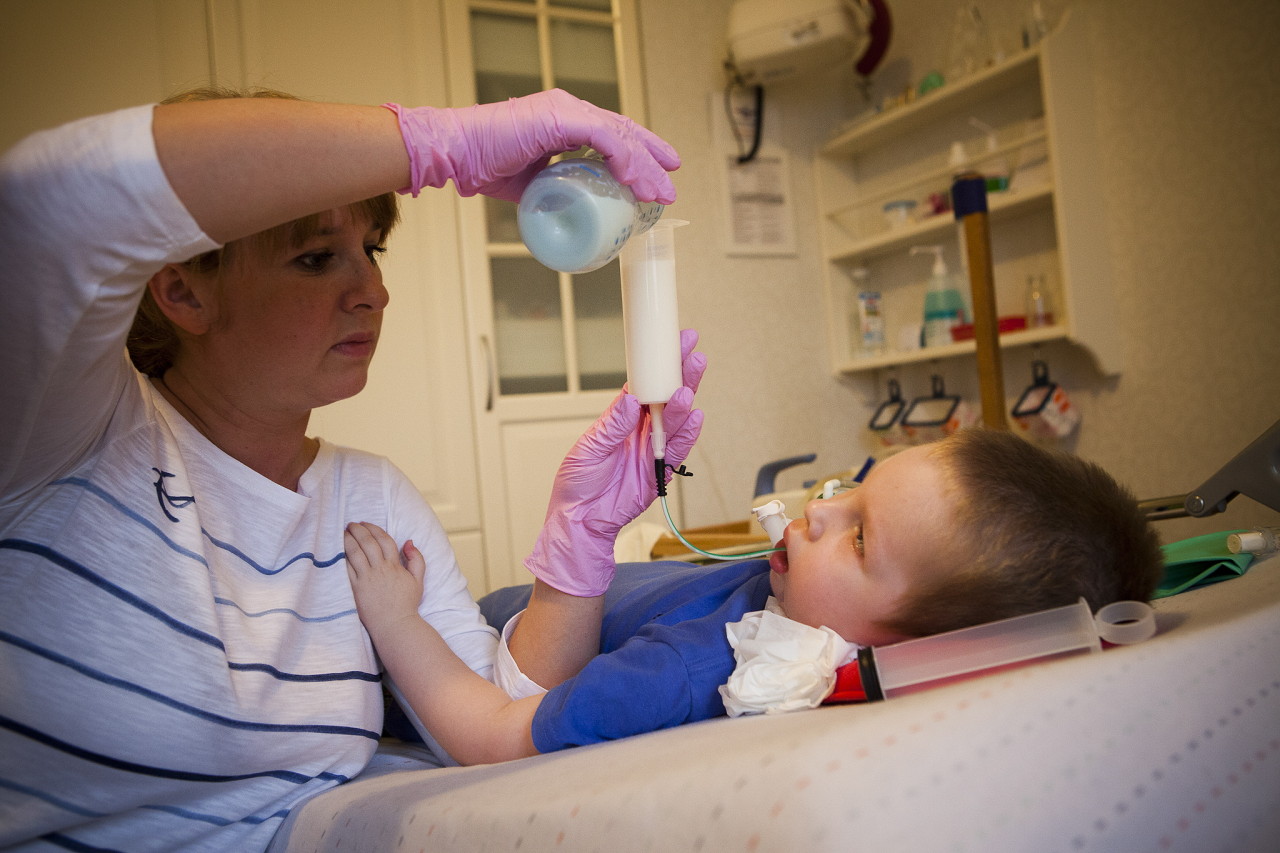

Mer informasjon om sykdommen forteller en spesialist i videoen nedenfor.