Спинална мишићна атрофија код деце
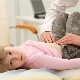
Спинална мишићна атрофија је тешка патологија која често прави једноставне радње, као што је ходање, седење, недоступност детету. Беба може бити лишена чак и такве природне могућности као самостално дисање. Врло је тешко предвидјети ову патологију, на крају крајева, не постоји ни посебан третман нити профилакса, али све зависи од облика болести и фактора, који лијек не може наћи објашњење.
Шта је ово?
Говорећи о спиналној мишићној атрофији, подразумева се не једна специфична болест, већ читава група болести под општом скраћеницом ЦМА. Сви они су наследни и повезани су са дегенерацијом нервних ћелија кичмене мождине, које су одговорне за моторичке функције.
Међу генетске патологије код деце спиналне мишићне атрофије заузимају водећа места у смислу учесталости ширења. И отприлике једна од 6 хиљада деце се рађа са тако страшном дијагнозом. У 50% случајева, дјеца не живе до двије године и умиру. Остатак живота је инвалидитет.

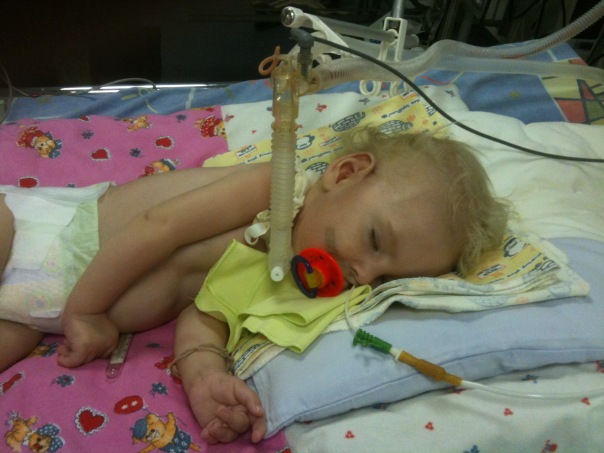
Проблем је, према генетичарима, много шири него што се чини из дате статистике.
Болест се развија због мутације одређених гена, а једна од њих је СМН1, који се сматра главним "кривцем" за патологију, у модификованом облику рецесивно присутног у сваком педесетом становнику планете. То значи да здрави родитељи, који чак и не схватају да су рецесивни носиоци мутираног гена, могу имати бебу са спиналном мишићном атрофијом.
Групу болести први је описао у 19. стољећу Гуидо Вердинг, чије је име касније проглашено једним од дјечјих сорти АГР.

Класификација
Најчешћи облик СМА код деце је проксималан. Представља га неколико врста болести, од којих све не постају видљиве одмах након рођења детета.
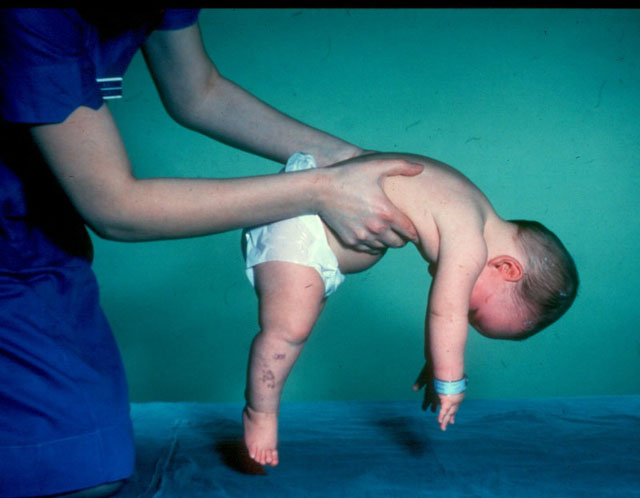
- Вердинг-Хоффманова болест - тип 1 МЦА, тешка обољења дојенчадикоја се манифестује у првих шест месеци живота детета. Прогнозе са својим најнеповољнијим, већина пацијената умире. Дете са СМА типа 1 не може ни стајати, ни седети, нити се самостално преврнути. Многе новорођенчади имају ослабљене рефлексе сисања и гутања. Често не постоји могућност спонтаног дисања или је тешко дисање.
- Атрофија Дубовица - ЦМА тип 2, лате инфант. Обично се јавља између шест мјесеци и годину и пол и касније. Дијете не може ходати, стајати, али је у стању да седи, храна није поремећена, он се носи са задатком гутања, сиса. Колико дуго ће беба да живи зависи од стања респираторних мишића.
- Атрофија Кугелберг-Веландер - ЦМА 3 врсте, инфантиле. Обично се налази у доби од једне и пол године, обично у двије године. Прогностички повољнији облик. Мали пацијенти могу стајати, сједити, кретати се, али осјећају велику слабост, па стога у већини случајева треба инвалидска колица, без којих им је нормална животна активност тешка.
- Кеннеди атрофија - ЦМА тип 4, булбоспиналнаја. Обично се сматра облик за одрасле, али повремено детектује код дјеце након 15 година. Утицај живота је ретко погођен, слабљење мишића се одвија полако, постепено, особа која је водила нормалан живот и сматрала се здравим, на крају постаје инвалид и губи способност самосталног кретања.

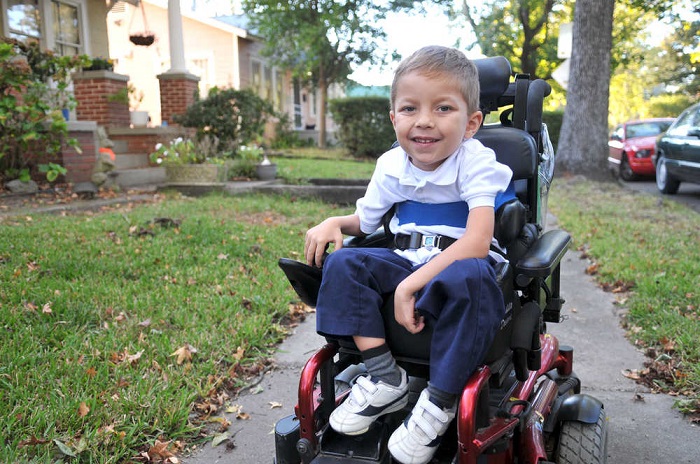

Више или мање позната атрофија Дуцхеннеа из прве руке и Вулпијанска атрофија је СМА "одрасли", први се обично открива након 18 година, а други након 20 година.
Код деце се не бележе само изоловани облици СМА, када ништа не смета осим дистрофије мишића, већ и комбиноване форме, када спинална атрофија није једина дијагноза, а дете има и друге генетске или урођене проблеме, као што су срчани и васкуларни дефекти, олигофренија.
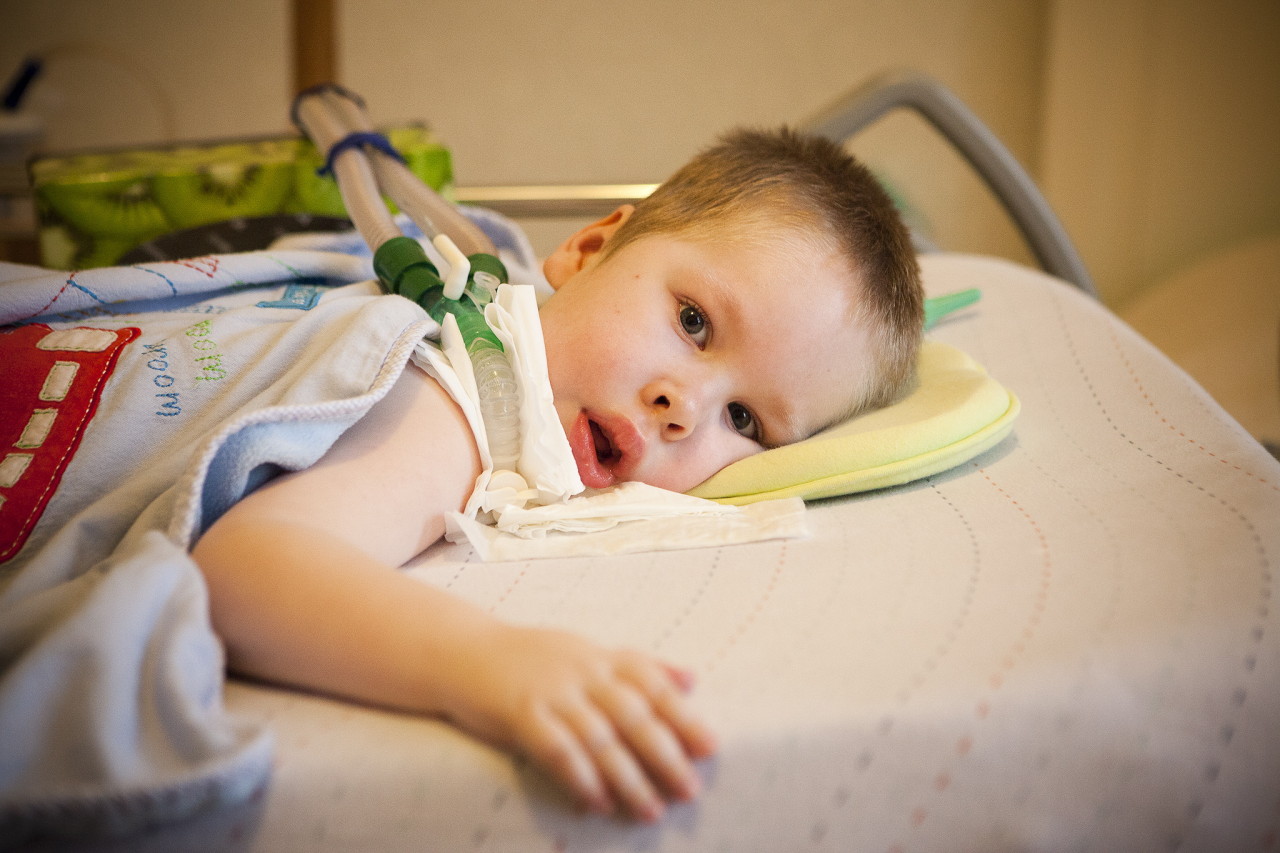
Разлози
Као што је већ поменуто, говоримо о генетској болести, па су разлози за њено појављивање поље претраживања генетичара. Дете наслеђује један од рецесивних гена на петом хромозому (то могу бити СМН, НАИП, Х4Ф5, БТФ2п44 гени).
Вероватноћа преношења таквог гена у потомство од носиоца је висока - 25%. Ако су и мама и тата скривени носиоци мутираног гена, онда је вероватноћа АГР код детета 50%. Оштећени абнормални ген спречава нормалну производњу СМН протеина и нервне ћелије одговорне за моторичке функције мишића у кичменој мождини почињу постепено да умиру. Процес њихове смрти наставља се и након рођења дјетета.
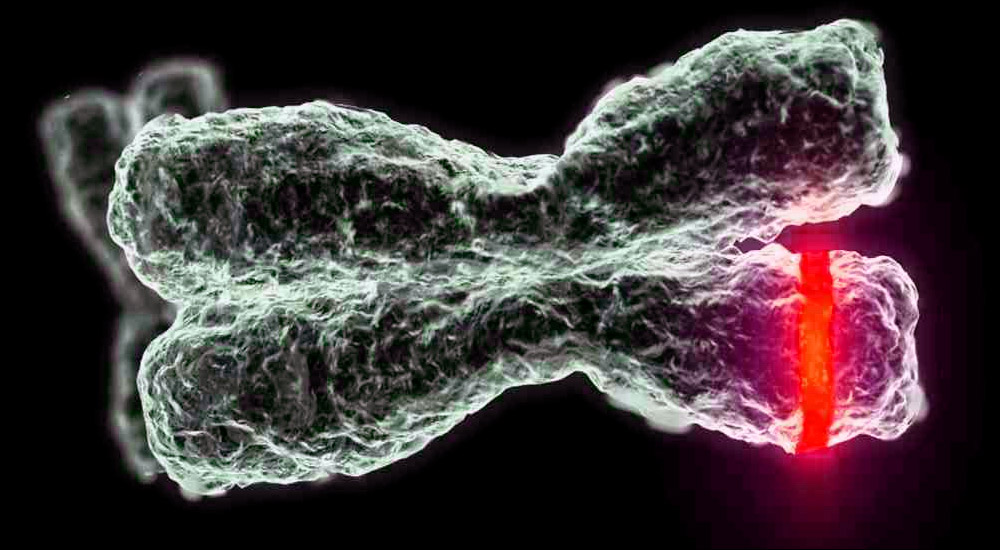

Манифестације
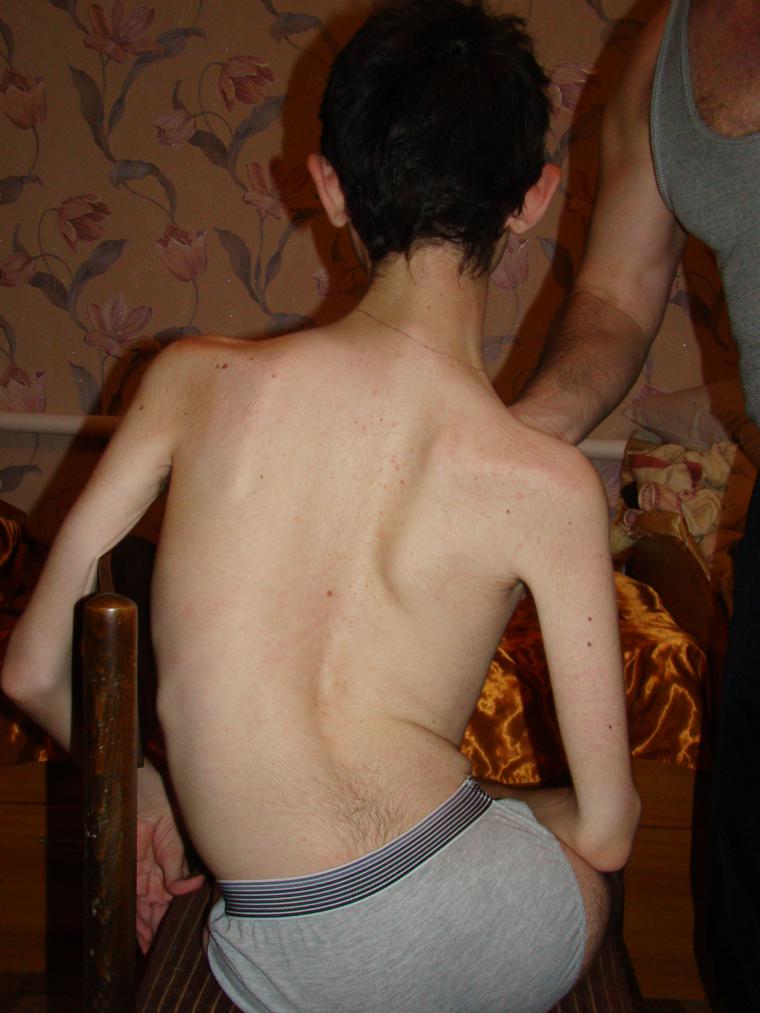
Симптоми зависе од врсте болести. Пошто разматрамо само четири типа деце, треба напоменути да су мишићна слабост и мишићна атрофија карактеристичне за све. У супротном, сваки тип има своју клиничку слику и карактеристичне особине.
- ЦМА тип 1 (Вердинг-Хоффманова атрофија) за детекцију чак и током трудноће. Лекар може да посумња на болест у фетусу веома споро. Али, да би се потврдила дијагноза у фази ношења детета је тешко, то се обично дешава након порода. Клинац са таквом атрофијом не може сам да држи главу, баца се са стране на страну, не седи. Готово увијек лежи на леђима, држање му је опуштено, не подиже ноге, не спаја их, не спушта дланове. У веома раној фази, могу се појавити велики проблеми да би се нахранило дијете, јер он гута, испада јако лоше или не ради. Већина дјеце умире прије своје двије године. Неки успевају да живе до седам или осам година, али атрофија се само погоршава. Обично се смрт догоди због недостатка срца, плућа, органа за варење.
- Тип ЦМА 2 (атрофија Дубовица) на рођењу се обично не детектује, јер дијете може дисати, прогутати храну, а тек након шест мјесеци напредује атрофија мишића. Ако се први симптоми јављају у годинама када је дете већ научило да стоји у кревету, онда знак за резање ногу, неразумна кап мрвица може бити светли знак. Постепено постаје тешко прогутати. Временом, дете почиње да му треба инвалидска колица.
- ЦМА тип 3 (амиотрофија Кугелберг-Веландер) може се појавити у било ком узрасту након 2 године живота. Дете које нормално расте и развија се постепено почиње жалити на слабост, обично на раменима, подлактицама. Како напредује, постаје му тешко трчати, ходати степеницама, чучати. Све зависи од бриге - неки задржавају способност самосталног кретања дуги низ година.
- ЦМА тип 4 (Кеннедијева атрофија) Појављује се само код мушких пацијената, јер се сматра повезаним са полним хромозомом Кс. Први знаци су слабост у подручју мишића бутина, а кранијални живци постепено захваћају. Болест напредује споро.

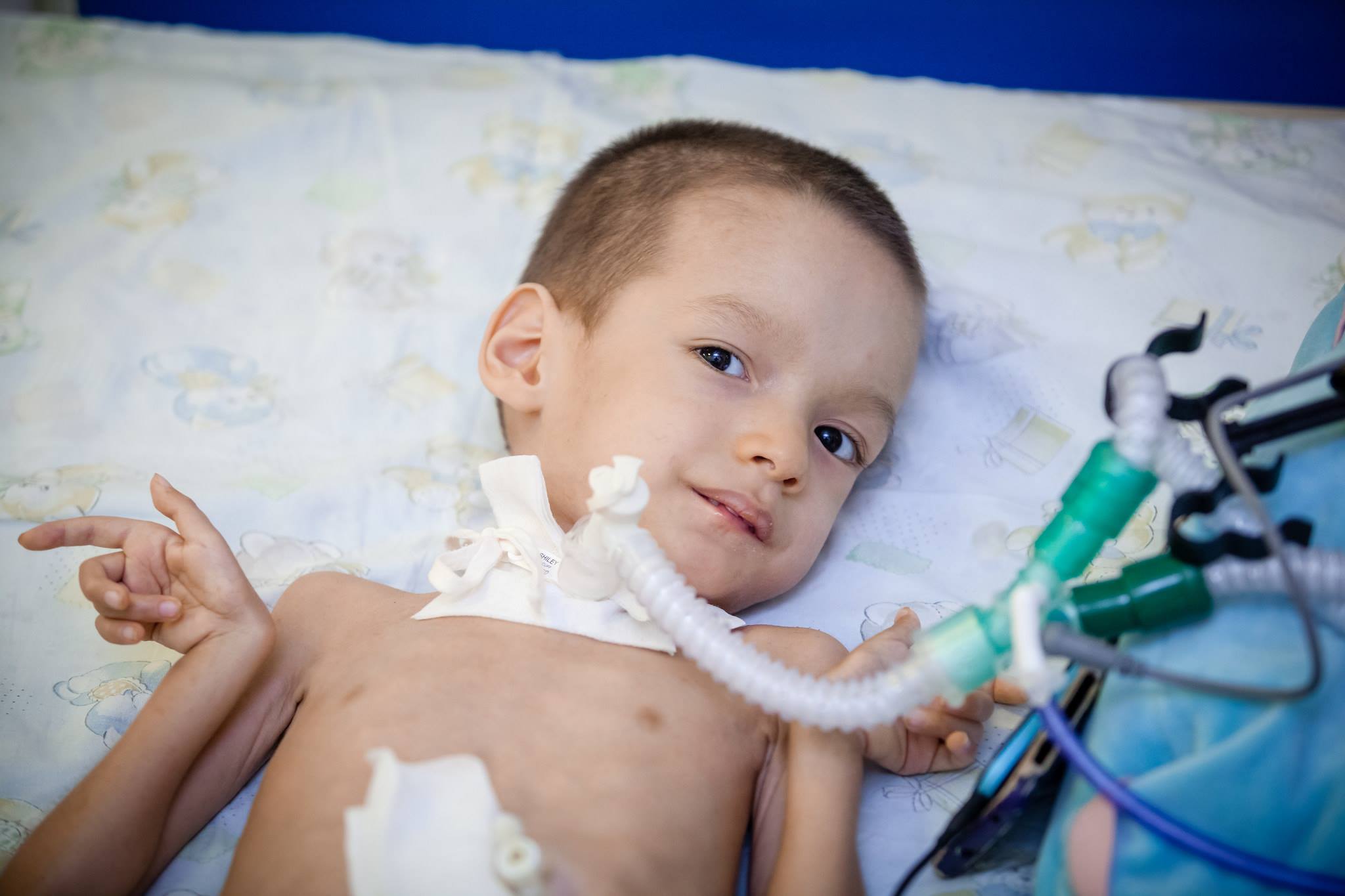
Третман
На жалост, данас медицинска наука не може да понуди методе и средства за лечење СМА. Таквих метода нема. Да би се одржале функције тела и максимизирао период док се дете не може кретати, лијекови као што су Прозерин, Оксазил. Оне смањују активност ензима који је способан за цијепање ацетилхолина, који преноси побудни пулс кроз влакна нервног система.
Такође препоручујемо систематска употреба лекова који побољшавају енергетски метаболизам на нивоу ћелија, витамини групе Б, ноотропни лекови, као и препарати калијум и никотинске киселине.
Дете са СМА приказана је високопротеинска дијетаАли недавне студије су показале да је улога дијете помало претјерана - нема доказа да висок садржај протеина у храни барем на неки начин утиче на стопу прогресије болести.
Али са калоријама треба бити опрезнији - због смањене мишићне активности, дијете може брзо добити додатне килограме.
Да би се продужио период више или мање испуњеног живота помоћи ће терапијска масажа, УХФ, електрофореза, вјежбе дисања за одржавање респираторних мишића, пливање. Препоручује се ношење носних спиналних или торакалних ортопедских помагала.

Више информација о болести говори специјалисту у видеу испод.